Tutorial for preparing and checking input data.¶
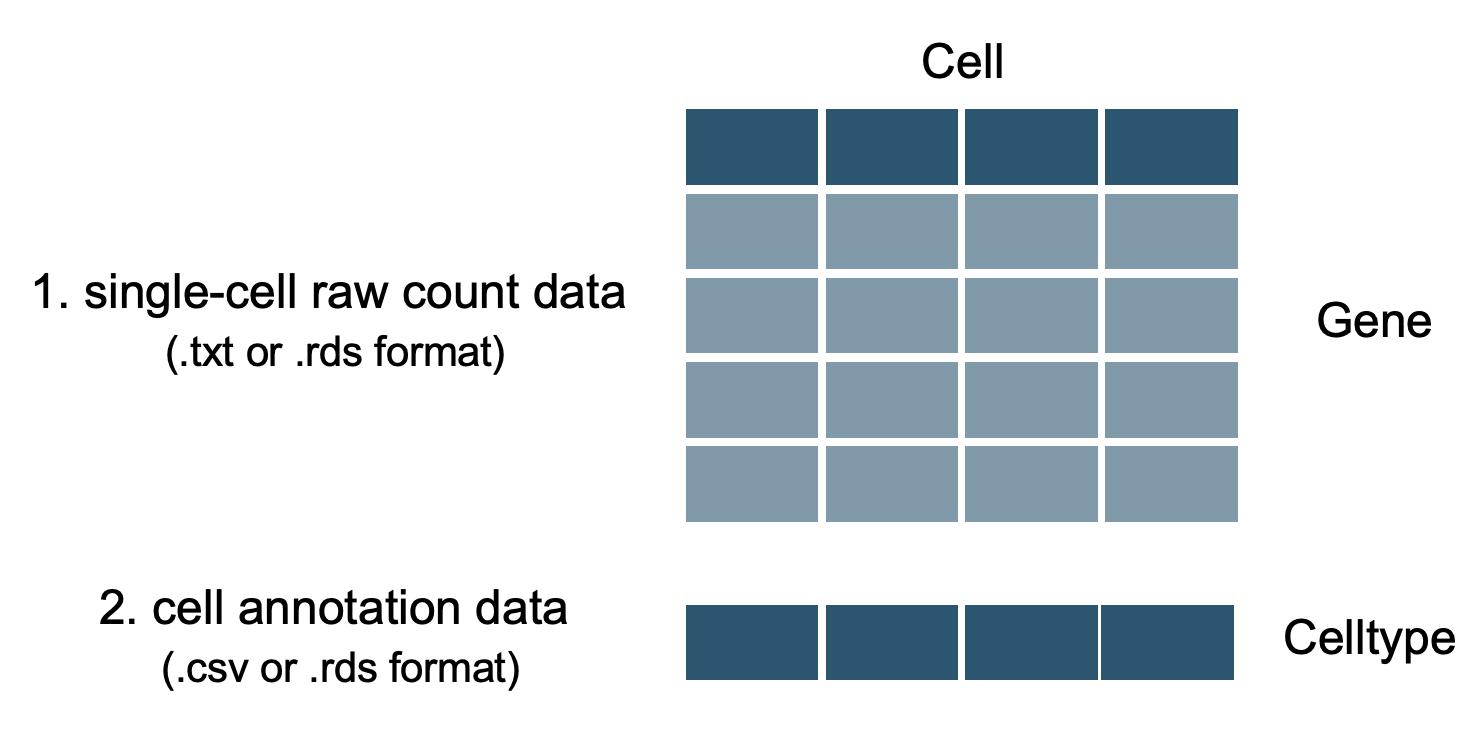
In [1]:
library(Seurat)
genecount <- as.matrix(read.table('txt_example.txt', header = TRUE, sep = ',')) ## ensure that the content in the txt file is comma-separated
genemeta <- read.csv('cell_anno_example.csv')
ser.obj <- CreateSeuratObject(genecount, meta.data=genemeta)
print(ser.obj)
Attaching SeuratObject Seurat v4 was just loaded with SeuratObject v5; disabling v5 assays and validation routines, and ensuring assays work in strict v3/v4 compatibility mode
An object of class Seurat 5000 features across 1308 samples within 1 assay Active assay: RNA (5000 features, 0 variable features) 2 layers present: counts, data
2. R data formatted file (.rds)¶
In [2]:
library(Seurat)
ser.obj <- readRDS('seurat_example.rds')
print(ser.obj)
An object of class Seurat 21005 features across 1308 samples within 1 assay Active assay: RNA (21005 features, 0 variable features) 2 layers present: counts, data
3. Anndata data file (.h5ad)¶
In [3]:
library(MuDataSeurat)
ser.obj <- ReadH5AD('h5ad_example.h5ad')
print(ser.obj)
An object of class Seurat 22315 features across 6022 samples within 1 assay Active assay: RNA (22315 features, 0 variable features) 2 layers present: counts, data
4. Seurat object data formatted file (.h5Seurat)¶
In [4]:
library(SeuratDisk)
ser.obj <- LoadH5Seurat('h5Seurat_example.h5Seurat')
print(ser.obj)
Registered S3 method overwritten by 'SeuratDisk': method from as.sparse.H5Group Seurat Validating h5Seurat file Initializing RNA with data Adding counts for RNA Adding miscellaneous information for RNA Adding command information Adding cell-level metadata Adding miscellaneous information Adding tool-specific results
An object of class Seurat 5000 features across 6022 samples within 1 assay Active assay: RNA (5000 features, 0 variable features) 2 layers present: counts, data
Check if a proper single-cell raw count data is available¶
In [5]:
print(ser.obj)
An object of class Seurat 5000 features across 6022 samples within 1 assay Active assay: RNA (5000 features, 0 variable features) 2 layers present: counts, data
In [6]:
print(GetAssayData(ser.obj, assay="RNA")[1:10, 1:10])
### select the first 10 rows and fist 10 columns as an example output
10 x 10 sparse Matrix of class "dgCMatrix"
[[ suppressing 10 column names ‘AdultLung_2.CTCGCAAACCTAGGCTGC’, ‘AdultLung_2.CTGTGTCTCCATACGTTG’, ‘AdultLung_2.CTCGCAACGTTGGTAATG’ ... ]]
SFTPC . . 10 38 . . 92 6 20 1 SFTPB . . 2 7 . . 24 1 5 4 FTL . 69 124 21 93 3 3 23 4 13 MT-RNR2 106 27 29 96 18 81 37 54 67 . SFTPA2 2 3 1 2 1 . 10 . 1 1 TMSB4X 30 61 103 21 81 37 19 19 31 . SCGB1A1 . . . . . . . . . . SFTPA1 1 . . 4 . . 14 2 1 . B2M 25 24 46 15 54 39 20 18 60 14 TPT1 15 8 8 11 4 9 23 7 9 1
Check if cell names in the gene expression matrix and the metadata are the same¶
In [7]:
print(all(colnames(ser.obj@assays$RNA) == ser.obj@meta.data$Cell_id))
[1] TRUE
Check if column 'Celltype' exists in metadata¶
In [8]:
print(any(grepl('Celltype', colnames(ser.obj@meta.data))))
[1] TRUE