OpenAnnotate facilitates studies of regulatory mechanism¶
OpenAnnotate has been successfully applied to studies of regulatory mechanism, including
- DeepTACT: redicting 3D chromatin contacts via bootstrapping deep learning. (Nucleic Acids Research 2019)
- DeepCAPE: a deep convolutional neural network for the accurate prediction of enhancers. (Genomics, Proteomics & Bioinformatics 2021)
- Gene co-opening network deciphers gene functional relationships. (Molecular BioSystems 2017)
- DeepHistone: a deep learning approach to predicting histone modifications. (BMC Genomics 2019)
- EpiFIT: functional interpretation of transcription factors based on combination of sequence and epigenetic information. (Quantitative Biology 2019)
Openness scores can be easily incorporated into machine learning models as the chromatin accessibility feature of genomic regions.
In this notebook, we provide a quick-start illustration of the usage of OpenAnnotate to accurately predict enhancers as in DeepCAPE with minor modifications for convenience.
We use a small dataset containing 166 human epithelial cell of esophagus enhancers as an example. The main steps are listed as follows:
- Download enhancers and generate potential negative samples
- Sample negative samples with the constraints on length and GC content
- Perform data augmentation and prepare BED files for OpenAnnotate
- Run OpenAnnotate and process the annotated openness scores
- Compile the DeepCAPE model with or without openness data
- Evaluate the prediction performance
Prepare positive and negative samples¶
Download the enhancers and skip the first line which contains introduction of the file.
!mkdir data
!wget http://enhancer.binf.ku.dk/presets/CL:0002252_epithelial_cell_of_esophagus_differentially_expressed_enhancers.bed -O ./data/epithelial_cell_of_esophagus_differentially_expressed_enhancers.bed
!tail ./data/epithelial_cell_of_esophagus_differentially_expressed_enhancers.bed -n +2 > ./data/epithelial_cell_of_esophagus_enhancers.bed
!head -5 ./data/epithelial_cell_of_esophagus_enhancers.bed
!wc -l ./data/epithelial_cell_of_esophagus_enhancers.bed
!mkdir output
To constrain the length of negative samples to be identically distributed as that of known enhancers, we prepare the potential negative samples of different lengths (as in gen_pool_neg.py) for the generation of chunked negative samples (as in gen_seq_data.py).
# Define functions
import hickle as hkl
import numpy as np
import os
acgt2num = {'A': 0,
'C': 1,
'G': 2,
'T': 3}
def seq2mat(seq):
seq = seq.upper()
h = 4
w = len(seq)
mat = np.zeros((h, w), dtype=bool) # True or False in mat
for i in range(w):
mat[acgt2num[seq[i]], i] = 1.
return mat.reshape((1, -1))
def checkseq(chrkey, start, end):
sequence = sequences[chrkey][start:end]
legal = ('n' not in sequence) and ('N' not in sequence)
return sequence, legal
def chunks(l, n, o):
"""
Yield successive n-sized chunks with o-sized overlap from l.
"""
return [l[i: i + n] for i in range(0, len(l), n-o)]
def cropseq_pool_neg(l, bedfile):
"""
Generate negative samples with specific length.
"""
stride = l
fin = open(GENOME_neg, 'r').readlines()
neg_samples = 0
f = open(bedfile, 'w')
for index in fin:
chrkey, startpos, endpos = index.strip().split('\t')
startpos = int(startpos)
endpos = int(endpos)
l_orig = endpos - startpos
if l_orig >= l:
chunks_ = chunks(range(startpos, endpos), l, l - stride)
for chunk in chunks_:
start = chunk[0]
end = chunk[-1] + 1
if (end - start) == l:
seq, legal = checkseq(chrkey, start, end)
if legal:
seq = seq.upper()
gc_content = 1.0 * (seq.count('G') + seq.count('C')) / l
f.write('%s\t%d\t%d\t%.3f\t.\t.\n' % (chrkey, start, end, gc_content))
neg_samples += 1
elif (end - start) < l:
break
print('Generated %d potential negative samples of length %d.'%(neg_samples, l))
return neg_samples
Input files can be downloaded here.
ENHANCER = './data/epithelial_cell_of_esophagus_enhancers.bed' # Downloaded enhancers
GENOME = './data/Chromosomes/' # FA files of chromosomes in hg19
GENOME_neg = './data/res_complement.bed' # Genome except the following regions
# 800k known enhancers data,33k known promoters data, 153k known lncpromoters data,
# 89k known lncrnas data, 963k known exons(CDS) data
!mkdir ./output/pool_neg
# Load whole genome sequence
chrs = list(range(1, 23))
chrs.extend(['X', 'Y'])
keys = ['chr' + str(x) for x in chrs]
if os.path.isfile('./output/sequences.hkl'):
print('Find corresponding hkl file')
sequences = hkl.load('./output/sequences.hkl')
else:
sequences = dict()
for i in range(24):
fa = open('%s%s.fa' % (GENOME, keys[i]), 'r')
sequence = fa.read().splitlines()[1:]
fa.close()
sequence = ''.join(sequence)
sequences[keys[i]]= sequence
hkl.dump(sequences, './output/sequences.hkl', 'w')
fin = open(ENHANCER, 'r')
length = []
for line in fin.readlines():
items = line.strip().split('\t')
length.append(int(items[2]) - int(items[1]))
fin.close()
for l in np.unique(length):
filename = './output/pool_neg/length_%d.bed'% l
neg_samples = cropseq_pool_neg(l, filename)
Generate negative samples and perform data augmentation¶
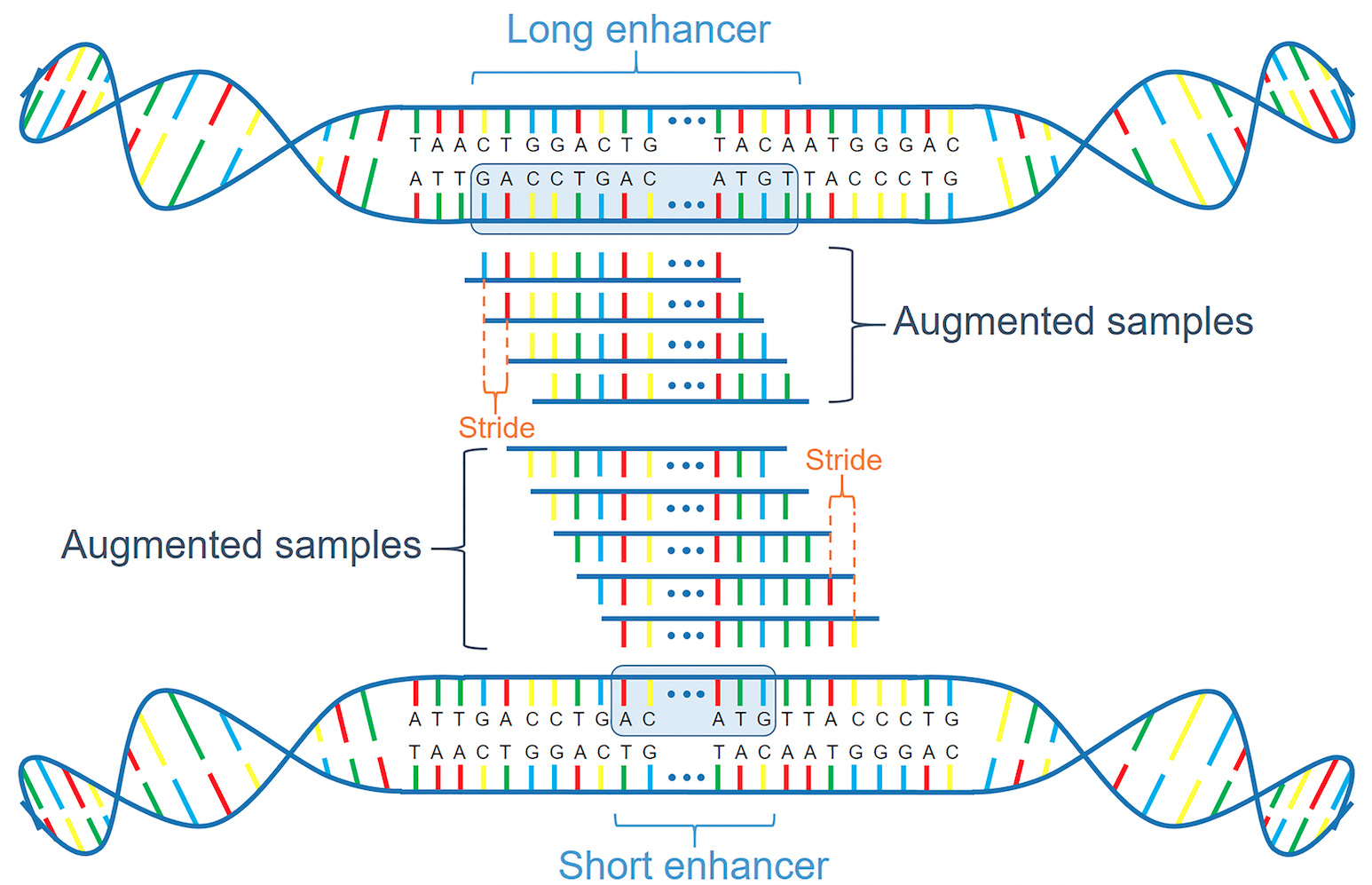
Augment positive and negative samples as in gen_seq_data.py. The outputs are BED files of samples in training and test sets to be submitted to OpenAnnotate, and
- train_X: augmented and one-hot encoded samples for training
- train_y: labels of augmented samples in training set
- test_X : augmented and one-hot encoded samples for test
- test_y : labels of augmented samples in test set
- test_pos_ids: original sample IDs of augmented positive samples in test set (to be used in evaluation)
- test_neg_ids: original sample IDs of augmented negative samples in test set (to be used in evaluation)
The original sample IDs are used in final prediction, that is, for each original test sample, the average predicted score of its augmented sequences is used as the final probability.
from sklearn.model_selection import KFold
all_neg = dict()
for key in keys:
all_neg[key] = list()
def loadindex(name, ratio):
"""
Load enhancers, and generate negative samples with the constraints on length and GC content
"""
print('Loading %s enhancer indexes...' % name)
file_name = './output/%s_index_ratio%d.hkl' % (name, ratio)
if os.path.isfile(file_name):
indexes, neg_indexes = hkl.load(file_name)
return indexes, neg_indexes
f = open(ENHANCER, 'r')
entries = f.readlines()
f.close()
indexes = list()
neg_indexes = list()
lens = list()
for i, entry in enumerate(entries):
chrkey, start, end = entry.split('\t')[:3]
lens.append(int(end) - int(start))
lens = np.array(lens)
bot = np.percentile(lens, 5)
top = np.percentile(lens, 95)
for i, entry in enumerate(entries):
chrkey, start, end = entry.split('\t')[:3]
start = int(start) - 1
end = int(end) - 1
seq, legal = checkseq(chrkey, start, end)
length = end - start
if legal and length > bot and length < top:
indexes.append(['%s%05d' % (name, i), chrkey, start, end])
seq = seq.upper()
pos_cont = 1.0 * (seq.count('G') + seq.count('C')) / length
f = open('./output/pool_neg/length_%d.bed' % length, 'r')
neg_entries = f.readlines()
f.close()
abs_cont = list()
for item in neg_entries:
neg_cont = item.split('\t')[3]
abs_cont.append(abs(pos_cont - float(neg_cont)))
sorted_ind = np.argsort(np.array(abs_cont)).tolist()
cnt = 0
for ind in sorted_ind:
neg_chrkey, neg_start, neg_end = neg_entries[ind].split('\t')[:3]
neg_start = int(neg_start) - 1
neg_end = int(neg_end) - 1
flag = 1
for item in all_neg[neg_chrkey]:
if (neg_start >= item[0] and neg_start <= item[1]) or (neg_end >= item[0] and neg_end <= item[1]) or (neg_end >= item[0] and neg_start <= item[0]) or (neg_end >= item[1] and neg_start <= item[1]) :
flag = 0
break
if flag:
cnt += 1
all_neg[neg_chrkey].append([neg_start, neg_end])
neg_indexes.append(['neg%05d_%d' % (i, cnt), neg_chrkey, neg_start, neg_end])
if cnt == ratio:
break
if cnt != ratio:
print('[Error! No enough negative samples!]')
print('Totally {0} enhancers and {1} negative samples.'.format(len(indexes), len(neg_indexes)))
hkl.dump([indexes, neg_indexes], file_name)
return [indexes, neg_indexes]
def train_test_split(indexes, neg_indexes, name, ratio):
"""
Split training and test sets.
(indexes: indexes to be split)
"""
print('Splitting the indexes into train and test parts...')
file_name = './output/%s_index_split_ratio%d.hkl' % (name, ratio)
if os.path.isfile(file_name):
print('Loading saved splitted indexes...')
return hkl.load(file_name)
n_samles = len(indexes)
kf = KFold(n_splits=5, shuffle=True, random_state=0)
kf.get_n_splits(range(n_samles))
allsplits = []
neg_allsplits =[]
for train, test in kf.split(range(n_samles)):
train_indexes = [indexes[i] for i in train]
test_indexes = [indexes[i] for i in test]
allsplits.append((train_indexes, test_indexes))
neg_train =[]
for i in train:
neg_train = neg_train + list(range(i*ratio, (i+1)*ratio))
neg_test =[]
for i in test:
neg_test = neg_test + list(range(i*ratio, (i+1)*ratio))
neg_train_indexes = [neg_indexes[i] for i in neg_train]
neg_test_indexes = [neg_indexes[i] for i in neg_test]
neg_allsplits.append((neg_train_indexes, neg_test_indexes))
print('Saving splitted indexes...')
hkl.dump([allsplits, neg_allsplits], file_name)
return [allsplits, neg_allsplits]
def to_bed_file(indexes, bedfile):
"""
Format indexes into bed file.
(indexes: indexes to be saved to bed files. bedfile: destination bed file name)
"""
print('Saving sequences into %s...' % bedfile)
f = open(bedfile, 'w')
for index in indexes:
if len(index) == 4:
[id, chrkey, start, end] = index
f.write('{0[1]}\t{0[2]}\t{0[3]}\t{0[0]}\t.\t.\n'.format(index))
elif len(index) == 5:
[id, chrkey, start, end, seq] = index
f.write('{0[1]}\t{0[2]}\t{0[3]}\t{0[0]}\t.\t.\n'.format(index))
else:
raise ValueError('index not in correct format!')
f.close()
def cropseq(indexes, l, stride):
"""
Generate chunked samples according to loaded indexes.
"""
enhancers = list()
for index in indexes:
try:
[sampleid, chrkey, startpos, endpos, _] = index
except:
[sampleid, chrkey, startpos, endpos] = index
l_orig = endpos - startpos
if l_orig < l:
for shift in range(0, l - l_orig, stride):
start = startpos - shift
end = start + l
seq, legal = checkseq(chrkey, start, end)
if legal and len(seq) == l:
enhancers.append((sampleid, chrkey, start, end, seq))
elif l_orig >= l:
chunks_ = chunks(list(range(startpos, endpos)), l, l - stride)
for chunk in chunks_:
start = chunk[0]
end = chunk[-1] + 1
if (end - start) == l:
seq, legal = checkseq(chrkey, start, end)
if legal and len(seq) == l:
enhancers.append((sampleid, chrkey, start, end, seq))
else:
break
print('Data augmentation: from {} indexes to {} samples'.format(len(indexes), len(enhancers)))
return enhancers
def generate_data(name, l, stride, ratio):
indexes, neg_indexes = loadindex(name, ratio)
allsplits, neg_allsplits = train_test_split(indexes, neg_indexes , name, ratio)
for i in range(5):
train_indexes, test_indexes = allsplits[i]
neg_train_indexes, neg_test_indexes = neg_allsplits[i]
TRAIN_POS_FA = './output/data1_%d/%s_train_stride%d_positive_%d.bed'% (ratio, name, stride, i)
TEST_POS_FA = './output/data1_%d/%s_test_stride%d_positive_%d.bed' % (ratio, name, stride, i)
TRAIN_NEG_FA = './output/data1_%d/%s_train_stride%d_negative_%d.bed'% (ratio, name, stride, i)
TEST_NEG_FA = './output/data1_%d/%s_test_stride%d_negative_%d.bed' % (ratio, name, stride, i)
train_pos = cropseq(train_indexes, l, stride) # data augmentation
test_pos = cropseq(test_indexes, l, stride) # data augmentation
to_bed_file(train_pos, TRAIN_POS_FA)
to_bed_file(test_pos, TEST_POS_FA)
train_neg = cropseq(neg_train_indexes, l, stride) # data augmentation
test_neg = cropseq(neg_test_indexes, l, stride) # data augmentation
to_bed_file(train_neg, TRAIN_NEG_FA)
to_bed_file(test_neg, TEST_NEG_FA)
train_y = [1] * len(train_pos) + [0] * len(train_neg)
train_y = np.array(train_y, dtype=bool)
train_X_pos = np.vstack([seq2mat(item[-1]) for item in train_pos])
train_X_neg = np.vstack([seq2mat(item[-1]) for item in train_neg])
train_X = np.vstack((train_X_pos, train_X_neg))
test_y = [1] * len(test_pos) + [0] * len(test_neg)
test_y = np.array(test_y, dtype=bool)
test_pos_ids = [item[0] for item in test_pos]
test_neg_ids = [item[0] for item in test_neg]
test_X_pos = np.vstack([seq2mat(item[-1]) for item in test_pos])
test_X_neg = np.vstack([seq2mat(item[-1]) for item in test_neg])
test_X = np.vstack((test_X_pos, test_X_neg))
hkl.dump((train_X, train_y), './output/data1_%d/%s_train_stride%d_%d_seq.hkl' % (ratio, name, stride, i), 'w')
hkl.dump((test_X, test_y), './output/data1_%d/%s_test_stride%d_%d_seq.hkl' % (ratio, name, stride, i), 'w')
hkl.dump(test_pos_ids, './output/data1_%d/%s_test_pos_ids_stride%d_%d.hkl' % (ratio, name, stride, i), 'w')
hkl.dump(test_neg_ids, './output/data1_%d/%s_test_neg_ids_stride%d_%d.hkl' % (ratio, name, stride, i), 'w')
break
!mkdir ./output/data1_5
# Here we consider the situation that the ratio of positive and negative samples is 1:5,
# the windows size for data augmentation is 300, and the stride for data augmentation is 1.
# We use only one fold of 5-fold cross validation to demonstrate.
name = 'epithelial_cell_of_esophagus'
ratio = 5
wdsize = 300
stride = 1
generate_data(name, wdsize, stride, ratio)
Submit to OpenAnnotate¶
OpenAnnotate extracts the first three columns and the sixth column (the chromosomes, starting sites, terminating sites and strands, respectively) separated by tabs for calculating openness scores.
Now we can submit the following saved BED files of training and test samples to OpenAnnotate on the 'Annotate' page iteratively:
- epithelial_cell_of_esophagus_train_stride1_positive_0.bed
- epithelial_cell_of_esophagus_test_stride1_positive_0.bed
- epithelial_cell_of_esophagus_train_stride1_negative_0.bed
- epithelial_cell_of_esophagus_test_stride1_negative_0.bed
Upload the file, choose 'DNase-seq (ENCODE)' and 'Homo sapiens (GRCh37/hg19)', specify the cell type of 'epithelial cell of esophagus', enable the option of Per-base pair annotation', and submit the task.
The four tasks will be finished within 10 minutes.
Download the annotation results by the links sent to mailbox or the online Download button.
# In this demo, you can directly use the following Download links for convenience.
!wget http://health.tsinghua.edu.cn/openness/anno/task/2020/1205/06540955/anno/head.txt.gz -O ./output/data1_5/epithelial_cell_of_esophagus_test_stride1_negative_0.head.txt.gz
!zcat ./output/data1_5/epithelial_cell_of_esophagus_test_stride1_negative_0.head.txt.gz
Each line in head.txt.gz contains the detailed information of corresponding column in the openness results.
Here we take the raw read openness (readopen.txt.gz) as an example.
# epithelial_cell_of_esophagus_train_stride1_positive_0.bed
!wget http://health.tsinghua.edu.cn/openness/anno/task/2020/1205/06540955/anno/readopen.txt.gz -O ./output/data1_5/epithelial_cell_of_esophagus_train_stride1_positive_0.readopen.txt.gz
# epithelial_cell_of_esophagus_test_stride1_positive_0.bed
!wget http://health.tsinghua.edu.cn/openness/anno/task/2020/1205/06135624/anno/readopen.txt.gz -O ./output/data1_5/epithelial_cell_of_esophagus_test_stride1_positive_0.readopen.txt.gz
# epithelial_cell_of_esophagus_train_stride1_negative_0.bed
!wget http://health.tsinghua.edu.cn/openness/anno/task/2020/1205/05423529/anno/readopen.txt.gz -O ./output/data1_5/epithelial_cell_of_esophagus_train_stride1_negative_0.readopen.txt.gz
# epithelial_cell_of_esophagus_test_stride1_negative_0.bed
!wget http://health.tsinghua.edu.cn/openness/anno/task/2020/1205/05354136/anno/readopen.txt.gz -O ./output/data1_5/epithelial_cell_of_esophagus_test_stride1_negative_0.readopen.txt.gz
Uncompress the files and load the results using Numpy. The first four columns consist of chromosomes, starting sites, terminating sites and strands, respectively. The two remaining columns corresponds to two DNase-seq experiments of epithelial cell of esophagus.
!gunzip ./output/data1_5/*.txt.gz
readopen = np.loadtxt('./output/data1_5/epithelial_cell_of_esophagus_train_stride1_positive_0.readopen.txt',dtype=str)
train_X_pos_open = readopen[:,4:].astype(float).reshape(-1, 2, 300, 1)
readopen = np.loadtxt('./output/data1_5/epithelial_cell_of_esophagus_test_stride1_positive_0.readopen.txt',dtype=str)
test_X_pos_open = readopen[:,4:].astype(float).reshape(-1, 2, 300, 1)
readopen = np.loadtxt('./output/data1_5/epithelial_cell_of_esophagus_train_stride1_negative_0.readopen.txt',dtype=str)
train_X_neg_open = readopen[:,4:].astype(float).reshape(-1, 2, 300, 1)
readopen = np.loadtxt('./output/data1_5/epithelial_cell_of_esophagus_test_stride1_negative_0.readopen.txt',dtype=str)
test_X_neg_open = readopen[:,4:].astype(float).reshape(-1, 2, 300, 1)
train_X_open = np.vstack((train_X_pos_open, train_X_neg_open))
test_X_open = np.vstack((test_X_pos_open, test_X_neg_open))
name = 'epithelial_cell_of_esophagus'
ratio = 5
stride = 1
kFold_i= 0
# Save the openness data
hkl.dump(train_X_open, './output/data1_%d/%s_train_stride%d_%d_open.hkl' % (ratio, name, stride, kFold_i), 'w')
hkl.dump(test_X_open, './output/data1_%d/%s_test_stride%d_%d_open.hkl' % (ratio, name, stride, kFold_i), 'w')
Compile the model and evaluate the performance¶
Load the openness data and the one-hot encoded sequence data.
import hickle as hkl
import numpy as np
name = 'epithelial_cell_of_esophagus'
ratio = 5
stride = 1
kFold_i= 0
train_X_open = hkl.load('./output/data1_%d/%s_train_stride%d_%d_open.hkl' % (ratio, name, stride, kFold_i))
test_X_open = hkl.load('./output/data1_%d/%s_test_stride%d_%d_open.hkl' % (ratio, name, stride, kFold_i))
train_X_seq, train_y = hkl.load('./output/data1_%d/%s_train_stride%d_%d_seq.hkl' % (ratio, name, stride, kFold_i))
test_X_seq, test_y = hkl.load('./output/data1_%d/%s_test_stride%d_%d_seq.hkl' % (ratio, name, stride, kFold_i))
test_pos_ids = hkl.load('./output/data1_%d/%s_test_pos_ids_stride%d_%d.hkl' % (ratio, name, stride, kFold_i))
test_neg_ids = hkl.load('./output/data1_%d/%s_test_neg_ids_stride%d_%d.hkl' % (ratio, name, stride, kFold_i))
train_X_seq = train_X_seq.reshape(-1, 4, 300, 1)
test_X_seq = test_X_seq.reshape(-1, 4, 300, 1)
train_y = np.array(train_y, dtype='uint8')
print(train_X_seq.shape, train_X_open.shape, train_y.shape, test_X_seq.shape, test_X_open.shape)
Shuffle the training samples.
indice = np.arange(train_y.shape[0])
np.random.shuffle(indice)
train_X_seq = train_X_seq[indice]
train_X_open = train_X_open[indice]
train_y = train_y[indice]
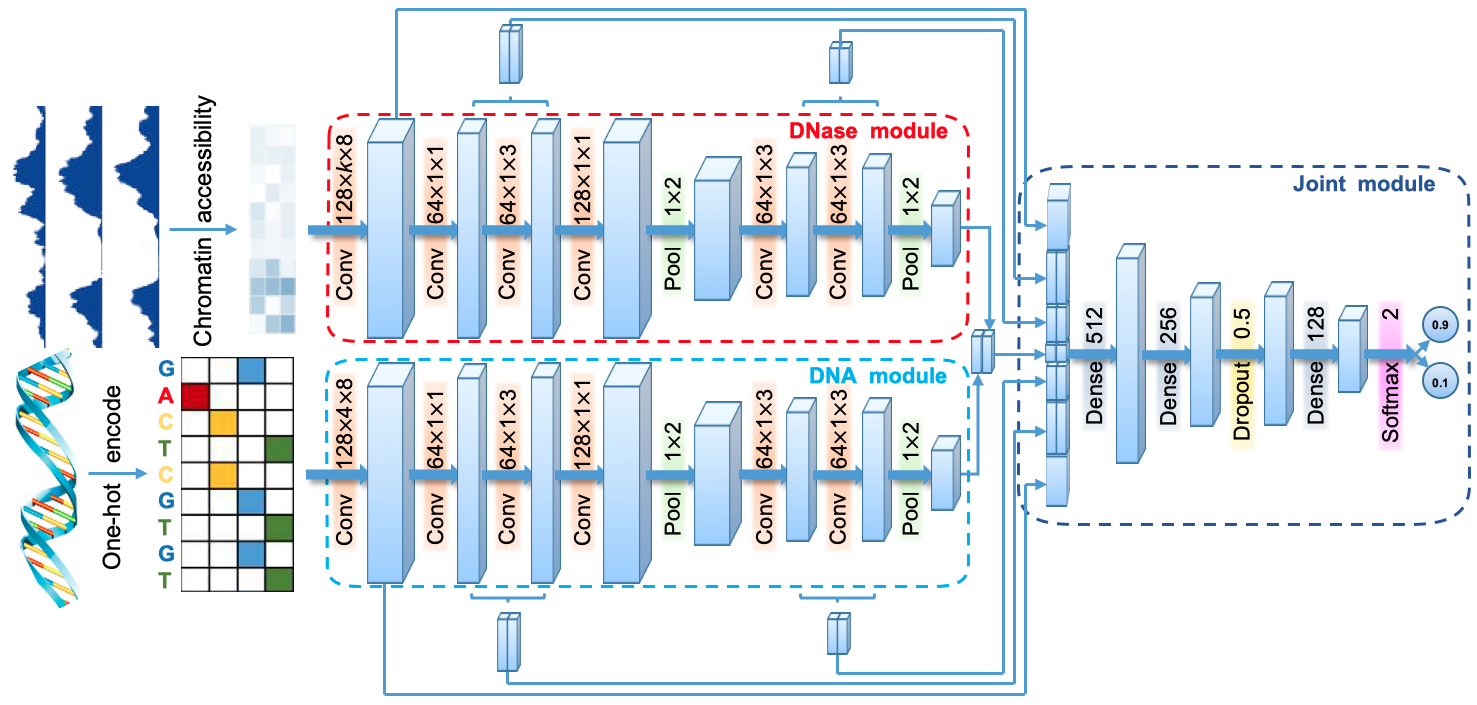
Note that for convenience, we donot use the auto-encoder and the pre-training stragy proposed in DeepCAPE.
import os
import keras as K
os.environ["CUDA_VISIBLE_DEVICES"] = '0'
import tensorflow as tf
import sys
from keras.models import Sequential, Model
from keras.layers import Input, Dense, Dropout, Flatten, Conv2D, MaxPooling2D, Concatenate
from keras.callbacks import EarlyStopping, ModelCheckpoint
from keras.optimizers import Adam
K.__version__
tf.__version__
Firstly, we build the 'seq only' model, a baseline method using only DNA sequence information.
input_seq = Input(shape=(4, 300, 1))
seq_conv1_ = Conv2D(128, (4, 8), activation='relu',padding='valid')
seq_conv1 = seq_conv1_(input_seq)
seq_conv2_ = Conv2D(64, (1, 1), activation='relu',padding='same')
seq_conv2 = seq_conv2_(seq_conv1)
seq_conv3_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv3 = seq_conv3_(seq_conv2)
seq_conv4_ = Conv2D(128, (1, 1), activation='relu',padding='same')
seq_conv4 = seq_conv4_(seq_conv3)
seq_pool1 = MaxPooling2D(pool_size=(1, 2))(seq_conv4)
seq_conv5_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv5 = seq_conv5_(seq_pool1)
seq_conv6_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv6 = seq_conv6_(seq_conv5)
#
seq_conv7_ = Conv2D(128, (1, 1), activation='relu',padding='same')
seq_conv7 = seq_conv7_(seq_conv6)
#
seq_pool2 = MaxPooling2D(pool_size=(1, 2))(seq_conv7)
merge_seq_conv2_conv3 = Concatenate(axis=-1)([seq_conv2, seq_conv3])
merge_seq_conv5_conv6 = Concatenate(axis=-1)([seq_conv5, seq_conv6])
x = Concatenate(axis=2)([seq_conv1, merge_seq_conv2_conv3, merge_seq_conv5_conv6, seq_pool2])
x = Flatten()(x)
dense1_ = Dense(512, activation='relu')
dense1 = dense1_(x)
dense2 = Dense(256, activation='relu')(dense1)
x = Dropout(0.5)(dense2)
dense3 = Dense(128, activation='relu')(x)
pred_output = Dense(1, activation='sigmoid')(dense3)
model = Model(inputs=[input_seq], outputs=[pred_output])
filename = './output/model-seq_only-data1_%d_%s_stride%d_%d.h5' % (ratio, name, stride, kFold_i)
early_stopping = EarlyStopping(monitor='val_loss', verbose=0, patience=3, mode='min')
save_best = ModelCheckpoint(filename, save_best_only=True, save_weights_only=True)
adam = Adam(lr=1e-4, beta_1=0.9, beta_2=0.999, epsilon=1e-08, decay=1e-6)
model.compile(loss='binary_crossentropy', optimizer=adam, metrics=['accuracy'])
model.fit(train_X_seq, train_y, batch_size=128, epochs=30, validation_split=0.1, callbacks=[early_stopping, save_best])
y_prob = model.predict(test_X_seq)
y_prob = y_prob[:, 0]
filename = './output/res-seq_only-data1_%d_%s_stride%d_%d.txt' % (ratio, name, stride, kFold_i)
f = open(filename, 'w')
f.write('id:\t predicted probability(ies)')
y_prob_new = []
temp = []
for i, id, lastid in zip(
range(len(test_pos_ids)),
test_pos_ids,
[None] + test_pos_ids[:-1]):
if id != lastid:
y_prob_new.append(temp)
temp = []
f.write('\n%s:\t' % id)
temp.append(y_prob[i])
f.write('%.4f\t' % y_prob[i])
y_prob_new.append(temp)
y_prob_pos = [sum(item)/len(item) for item in y_prob_new[1:]]
n_pos = len(y_prob_pos)
y_prob_new = []
temp = []
for i, id, lastid in zip(
range(len(test_pos_ids),len(test_pos_ids)+len(test_neg_ids)),
test_neg_ids,
[None] + test_neg_ids[:-1]):
if id != lastid:
y_prob_new.append(temp)
temp = []
f.write('\n%s:\t' % id)
temp.append(y_prob[i])
f.write('%.4f\t' % y_prob[i])
y_prob_new.append(temp)
y_prob_neg = [sum(item)/len(item) for item in y_prob_new[1:]]
n_neg = len(y_prob_neg)
y_prob_new = y_prob_pos + y_prob_neg
y_test = [1] * n_pos + [0] * n_neg
print('test_pos_num:',n_pos,'test_neg_num:',n_neg)
y_pred = np.array(np.array(y_prob_new) > 0.5, dtype='int32')
The prediction performance is evaluated by the area under the precision-recall curve (auPRC) and the area under the receiver operating characteristic curve (auROC).
from sklearn import metrics
auROC = metrics.roc_auc_score(y_test, y_prob_new)
print('auROC = {}'.format(auROC))
auPR = metrics.average_precision_score(y_test, y_prob_new)
print('auPR = {}'.format(auPR))
Next, we build the DeepCAPE model, which additionally incorporates the information of chromatin accessibility.
input_seq = Input(shape=(4, 300, 1))
input_open = Input(shape=(2, 300, 1))
seq_conv1_ = Conv2D(128, (4, 8), activation='relu',padding='valid')
seq_conv1 = seq_conv1_(input_seq)
seq_conv2_ = Conv2D(64, (1, 1), activation='relu',padding='same')
seq_conv2 = seq_conv2_(seq_conv1)
seq_conv3_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv3 = seq_conv3_(seq_conv2)
seq_conv4_ = Conv2D(128, (1, 1), activation='relu',padding='same')
seq_conv4 = seq_conv4_(seq_conv3)
seq_pool1 = MaxPooling2D(pool_size=(1, 2))(seq_conv4)
seq_conv5_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv5 = seq_conv5_(seq_pool1)
seq_conv6_ = Conv2D(64, (1, 3), activation='relu',padding='same')
seq_conv6 = seq_conv6_(seq_conv5)
seq_pool2 = MaxPooling2D(pool_size=(1, 2))(seq_conv6)
open_conv1_ = Conv2D(128, (2, 8), activation='relu',padding='valid')
open_conv1 = open_conv1_(input_open)
open_conv2_ = Conv2D(64, (1, 1), activation='relu',padding='same')
open_conv2 = open_conv2_(open_conv1)
open_conv3_ = Conv2D(64, (1, 3), activation='relu',padding='same')
open_conv3 = open_conv3_(open_conv2)
open_conv4_ = Conv2D(128, (1, 1), activation='relu',padding='same')
open_conv4 = open_conv4_(open_conv3)
open_pool1 = MaxPooling2D(pool_size=(1, 2))(open_conv4)
open_conv5_ = Conv2D(64, (1, 3), activation='relu',padding='same')
open_conv5 = open_conv5_(open_pool1)
open_conv6_ = Conv2D(64, (1, 3), activation='relu',padding='same')
open_conv6 = open_conv6_(open_conv5)
open_pool2 = MaxPooling2D(pool_size=(1, 2))(open_conv6)
merge_seq_conv2_conv3 = Concatenate(axis=-1)([seq_conv2, seq_conv3])
merge_seq_conv5_conv6 = Concatenate(axis=-1)([seq_conv5, seq_conv6])
merge_open_conv2_conv3 = Concatenate(axis=-1)([open_conv2, open_conv3])
merge_open_conv5_conv6 = Concatenate(axis=-1)([open_conv5, open_conv6])
merge_pool2 = Concatenate(axis=-1)([seq_pool2, open_pool2])
x = Concatenate(axis=2)([seq_conv1, merge_seq_conv2_conv3, merge_seq_conv5_conv6, merge_pool2, merge_open_conv5_conv6, merge_open_conv2_conv3, open_conv1])
x = Flatten()(x)
dense1_ = Dense(512, activation='relu')
dense1 = dense1_(x)
dense2 = Dense(256, activation='relu')(dense1)
x = Dropout(0.5)(dense2)
dense3 = Dense(128, activation='relu')(x)
pred_output = Dense(1, activation='sigmoid')(dense3)
model = Model(inputs=[input_seq, input_open], outputs=[pred_output])
filename = './output/model-seq_open-data1_%d_%s_stride%d_%d.h5' % (ratio, name, stride, kFold_i)
early_stopping = EarlyStopping(monitor='val_loss', verbose=0, patience=3, mode='min')
save_best = ModelCheckpoint(filename, save_best_only=True, save_weights_only=True)
adam = Adam(lr=1e-4, beta_1=0.9, beta_2=0.999, epsilon=1e-08, decay=1e-6)
model.compile(loss='binary_crossentropy', optimizer=adam, metrics=['accuracy'])
model.fit([train_X_seq, train_X_open], train_y, batch_size=128, epochs=30, validation_split=0.1, callbacks=[early_stopping, save_best])
y_prob = model.predict([test_X_seq, test_X_open])
y_prob = y_prob[:, 0]
filename = './output/res-seq_open-data1_%d_%s_stride%d_%d.txt' % (ratio, name, stride, kFold_i)
f = open(filename, 'w')
f.write('id:\t predicted probability(ies)')
y_prob_new = []
temp = []
for i, id, lastid in zip(
range(len(test_pos_ids)),
test_pos_ids,
[None] + test_pos_ids[:-1]):
if id != lastid:
y_prob_new.append(temp)
temp = []
f.write('\n%s:\t' % id)
temp.append(y_prob[i])
f.write('%.4f\t' % y_prob[i])
y_prob_new.append(temp)
y_prob_pos = [sum(item)/len(item) for item in y_prob_new[1:]]
n_pos = len(y_prob_pos)
y_prob_new = []
temp = []
for i, id, lastid in zip(
range(len(test_pos_ids),len(test_pos_ids)+len(test_neg_ids)),
test_neg_ids,
[None] + test_neg_ids[:-1]):
if id != lastid:
y_prob_new.append(temp)
temp = []
f.write('\n%s:\t' % id)
temp.append(y_prob[i])
f.write('%.4f\t' % y_prob[i])
y_prob_new.append(temp)
y_prob_neg = [sum(item)/len(item) for item in y_prob_new[1:]]
n_neg = len(y_prob_neg)
y_prob_new = y_prob_pos + y_prob_neg
y_test = [1] * n_pos + [0] * n_neg
print('test_pos_num:',n_pos,'test_neg_num:',n_neg)
y_pred = np.array(np.array(y_prob_new) > 0.5, dtype='int32')
from sklearn import metrics
auROC = metrics.roc_auc_score(y_test, y_prob_new)
print('auROC = {}'.format(auROC))
auPR = metrics.average_precision_score(y_test, y_prob_new)
print('auPR = {}'.format(auPR))
The significant improvement indicates that the chromatin accessibility data, which can be easily and efficiently obtained by OpenAnnotate, is capable of introducing cell line-specific information to the model, boosting the performance, and hence facilitating studies of regulatory mechanism.